Zespół Pfeiffera: przyczyny, objawy, leczenie
Zespół Pfayffera występuje niezwykle rzadkochoroba genetyczna występująca średnio u jednego na 100 000 noworodków. Równie często dotyka chłopców i dziewcząt. Głównym objawem choroby jest wczesne zespolenie kości czaszki w procesie embriogenezy, z powodu którego mózg nie może później normalnie się rozwijać.
Informacje ogólne
Choroba została odkryta przez słynnego Niemcagenetyk Rudolf Pfeiffer. W 1964 roku opisał objawy nieznanej wcześniej choroby, której głównymi objawami były nieprawidłowości w budowie czaszki i zespolenie palców na podeszwach stóp lub dłoni (syndaktylia). Zespół ten zaobserwowano u ośmiu pacjentów z tej samej rodziny, co wskazywało na genetyczny charakter choroby. Dalsze badania wykazały, że zespół Pfeiffera jest przenoszony przez autosomalny typ dominujący.
Ultradźwięki mogą wykryć tę chorobę u płodu już w drugim trymestrze ciąży. Na USG widoczne są nieprawidłowości w rozwoju czaszki, narządów wewnętrznych i kończyn.
Objawy
W 1993 roku amerykański genetyk Michael CohenZaproponował klasyfikację zespołu Pfeiffera na trzy typy w zależności od nasilenia objawów. Teraz jego klasyfikacja jest ogólnie akceptowana i szeroko stosowana w literaturze medycznej.
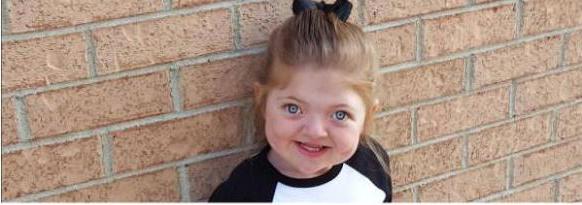
Objawy zespołu Pfayffera typu 1 obejmującraniosynostosis - stan, w którym jeden lub więcej szwów czaszkowych przedwcześnie się łączy, tworząc holistyczną kość. W wyniku tego obserwuje się zmiany kształtu czaszki i nienaturalne rysy twarzy - hiperteloryzm, niskie osadzone uszy. Może wystąpić pogorszenie wzroku, zwiększenie ciśnienia wewnątrzczaszkowego, rozszczep podniebienia. U 50% pacjentów zanotowano ubytek słuchu. Większość osób z tym typem choroby ma normalny poziom inteligencji i nie ma zaburzeń neurologicznych.
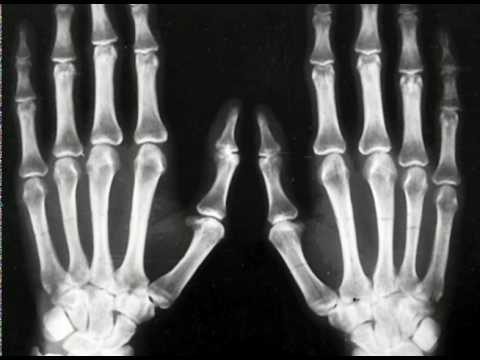
U dzieci z zespołem Pfayffera typu 1, zewnętrzneznaki, takie jak szerokie krótkie palce na rękach i nogach, są obserwowane prawie zawsze. Syndactyly jest możliwym, ale nie koniecznym objawem tego typu patologii.

Ten typ zespołu dziedziczy się autosomalnie dominująco.
Poniżej znajduje się zdjęcie zespołu Pfeiffera. Średnia długość życia w Rosji dla osób cierpiących na tę chorobę przekracza 60 lat.
W przypadku choroby typu 2 czaszka charakteryzuje się:forma "trefoil", połączenie kręgów, syndactyly i przesunięcie gałek ocznych (proptosis). Od pierwszych dni życia pojawiają się poważne zaburzenia neurologiczne.
Trzeci rodzaj choroby charakteryzuje się jeszcze bardziejwczesna kraniosynostoza. Czaszka jest nienormalnie wydłużona i nie tworzy "koniczyny". Występują nieprawidłowości zębów, hiperteloryzm, upośledzenie umysłowe, wady wewnętrzne narządów.
Żywotność w typie 2 i 3choroba waha się od kilku dni do kilku miesięcy - wady narządów wewnętrznych i zaburzenia neurologiczne nie są zgodne z życiem. Mutacje odpowiedzialne za te dwa typy zespołu Pfeiffera występują sporadycznie i nie są genetycznie dziedziczone.
Przyczyny
Zespół Pfayffera jest związany z mutacjami receptora 1czynnik wzrostu fibroblastów (FGFR1) na chromosomie 8 lub czynnik wzrostu fibroblastów receptora 2 (FGFR2) na chromosomie 10 genów, które wpływają na mutację odpowiedzialną za normalnego wzrostu fibroblastów, która odgrywa kluczową rolę w rozwoju kości ciała.

Leczenie
Współczesna medycyna nie pozwala jeszcze na eliminacjęmutacje w ludzkim genomie, nawet jeśli wiadomo, w których genach była pierestrojka. Dlatego w przypadku zespołu Pfeiffera, podobnie jak w przypadku większości innych chorób genetycznych, leczenie ma charakter objawowy. U pacjentów z typem 2 i typem 3 rokowanie jest niekorzystne: nie można skorygować wielu nieprawidłowości rozwojowych ani medycznie, ani chirurgicznie.
Zespół Pfayffera typu 1 jest zgodny z życiem. Główny objaw choroby - przedwczesne zespolenie kości czaszki - można skorygować chirurgicznie. Operacja jest zalecana dla dzieci w wieku do trzech miesięcy.

Ponadto interwencja chirurgiczna jest skuteczna w przypadku prostego syndaktylu. Leczenie najlepiej rozpocząć w wieku 18-24 miesięcy, podczas aktywnego wzrostu palców rąk i nóg.